- Synthesis and Reactivity of Silicon Transition Metal Complexes (Publication Series 1 - 60)
Recent studies have focused on the
design of model compounds to conceive the effect of diverse metal
fragments on the Si-H mojety. In this context the metallo-silanes 1 -5 containing one, two or three Si-bound metal fragments as well as metallodisilanes 6
have been made available by convenient routes.These "special" silanes
show extraordinary activation of the SiH-bonds by the transition metal
fragment which guarantees smooth "transition metallation" of the
silicon to afford inter alia the cluster compounds 7.

Interestingly the metallo-silanes can be oxygenated by using dimethyldioxyrane or H2O2/urea in the presence of MeReO3 as an oxygen transfer reagent.
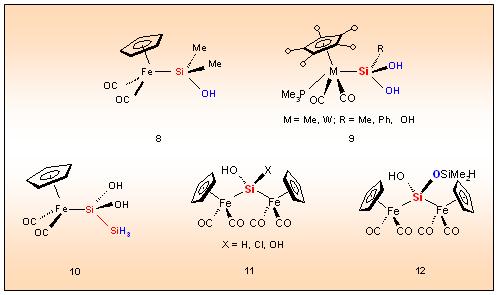
According to this method the metal fragment substituted silanols, silanediols or silanetriols 8, 9 are accessible. An interesting aspect refers to the regiospecifity of the oxygen transfer, which
guarantees the formation of unusual ligands at the metal centre like Si(OH)2SiH3 in 10 starting with the metallo-disilanes 6.
This product indicates the extraordinary hydride activity of SiH-bonds
directly connected to the metal - a consequence of the electron
releasing effect of the transition metal. Moreover bis(metallo)silanols
11
can be realized showing for the first time hydroxysilylenes bridging
two metal centres. Regiospecifity of the oxygenation process is also
valid for the bis-metallated species as is proved by the controlled
conversion of Cp(OC)2Fe-Si(H)(OSiMe2H)Fe(CO)2Cp, characterized by two types of SiH bonds, to the bis(metallo)siloxanol 12.
All metal fragment substituted silanols show unusual high stability
with respect to self condensation offering the possibility to perform
controlled condensation reaction with chlorosilanes, alkyls of the
group 13 elements and metal halides of Ti, Zr and Hf respectively, as
is demonstrated by the metallo-heterosiloxanes 13 and 14.
These results characterize metallo-silanols as attractive starting
materials for connecting the late metals Fe, Ru, Mo, W with Al, Ga, In
or the early metals Ti, Zr, Hf via SiO-units.
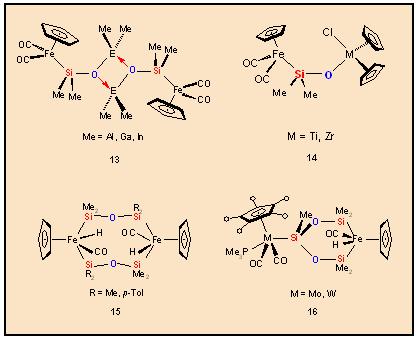
The SiH-functionalized metallosiloxanes deriving from 8 or 9
undergo facile oxidative addition of the Si-H bonds under photochemical
conditions to yield siloxanediyl bridged dinuclear complexes of the
type 15 and 16.
Recent efforts have been focused on metallo-silanols, in which the
"transition metal effect" is reduced by introducing a spacer unit. Such
metallo-silanols promiss elegant access to metal-substituted oligo- and
polysiloxanes via controlled self-condensation, an approach that has been realized for the first time with the metallo-silanols 17 having the metal and the silanol separated by a methylene spacer.
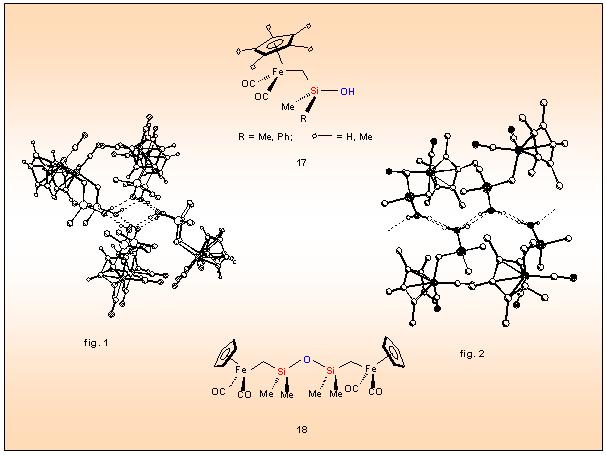
The Cp(OC)2Fe-CH2-substituted silanols 17, showing in the solid state extensive hydrogen bonding (fig. 1 and 2), exhibit enhanced reactivity towards self-condensation compared to the analogous Fe-Si systems. As a consequence, 17 exerts already at room temperature quantitative conversion to the 1,3-bis(ferriomethyl)disiloxane 18.
- Transition Metal Substituted Phosphines, Arsines and Stibines (Publication Series 1- 60)
These
studies made avalable a vast number of transition metal fragment
substituted phophines, arsines, stibines and even bismutines of the the
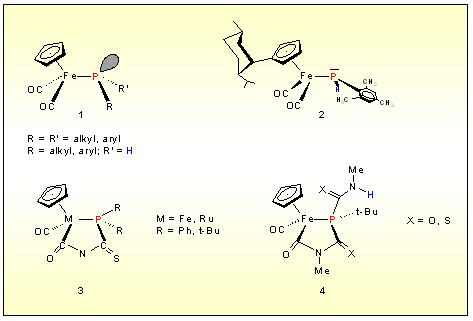
metals Cr, Mo, W, Fe,and Ru. Half sandwich complexes of the type 1 (R=R´= i-Pr) and 2
offered for the first to determine the influence of transition metals
on the phosphorus inversion, resulting in asignifant lowering of the
barrier. On the other hand nucleophilicity of the phosphorus is
considerably raised and gurantees spontaneous coupling with diverse
multiple bonding system to yield diastereoselctively products like 3. In the case of secondary metallo-phosphines (R´= H) an additional insertion process is observed to generate 4.
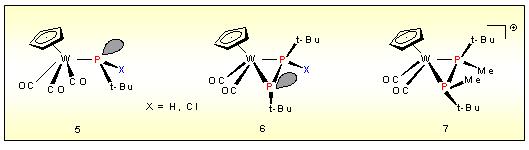
Functionalized Metallo-phosphines like 5
act as a source of phophinidines as well as the trapping reagent for
this short-lived species after transformation to a M=P doubled bond
complex via decarbonylation. The P-P coupling product 6
is produced in a stereochemically controlled manner. Further
interacction with elctrophiles or chalcogens affords diphosphine
ligands which can be removed from the metal under appropriate
conditions.
In
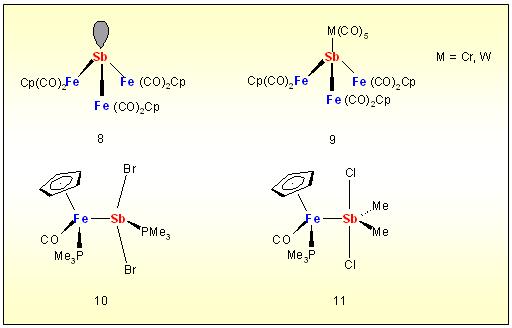
context with the heavier group 15 elements full transition metalliation
of antimony has been realized for the first time as well as the
coordination of the tris(ferrio)stibine 1 to give 2. First proof has been also given for Lewis acitity of metallo-dihalostibines by the formstion of 3 after phosphine addition and the existence of the transition metal substituted pentavalent antimony species 4.
- Transion Metal Assisted Hydrophosphination
Phosphines
attract increasing interest due to their use as ligands in
catalytically active transition metal complexes especially for
entantioselective synthesis. Our contributions to this topic refer to
the metal mediated synthesis of multifunctionalized and/or chiral
phosphines.
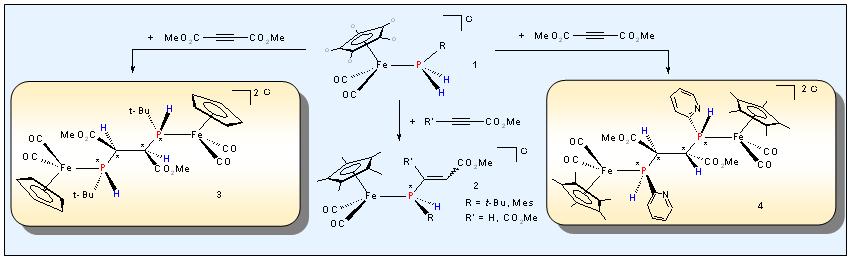
Previous studies concerning the
coupling reactions of metallo-phosphines with organic multiple bond
systems (see Section II) suggest an advantageous modification starting
with primary phosphine complexes of the chromium, manganese and iron
group metals e. g. 1 ( R' = alkyl, aryl,
py) and generating the corresponding phosphanido metal species via
deprotonation as a short-lived intermediate. Prominent examples arise
from the stepwise diastereoselctive hydrophosphination of electron
deficient alkynes resulting in the succesive formation of the secondary
vinylphophine complex 2 and the dinuclear iron complexes 3 and 4 bearing novel types of P-H-functionalized bis-phosphinoethane ligands as a bridging unit.
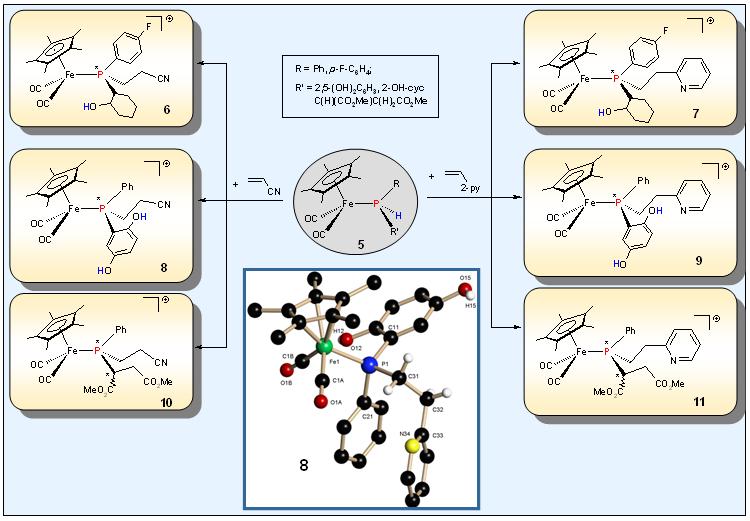
The introduction of a further functional groups into the secondary phosphine complexes 5
obtained by hydrophosphination involving the C=O- or C=C-bond of
quinones or olefins as well as the epoxy unit of cyclohexeneoxide can
be achieved by insertion of acrylonitrile or vinyl pyridine, which
leads to the regio- and diastereoselctive formation of the tertiary
phosphine complexes 6 - 11 . The
phosphorus ligand in these complexes offers an additional nitrogen
donor site, implying hybrid character appropriate for the coordination
of “hard” metals.
In
general the multifunctionalized chiral phosphines can released from the
metal under mild conditions and used in conventional organophosphorus
synthesis. Ongoing studies concentrate on the enantioselective
generation of the novel phosphines using chiral metal fragments or/ and
chiral phosphorus donors.

Especially P-H-functionalized phosphenium complexes Cp(OC)2W=P(H)R (1 ),
which offer a tremendous reactivity potential, create serious problems
concerning isolation due to insufficient shielding of the M=P bond.
While the Mes-compound shows controlled dimerisation to the
phosphinidene-bridged dinuclear complex 2 , the Ph-derivative undergoes [2+2]-cycloaddition to give 3. The tert -butyl-complex suffers immediate decomposition just below room temperature .
Only the supermesityl phosphenium complex Cp(OC)2W=P(H) s -Mes (4) could be isolated and structurally identified.
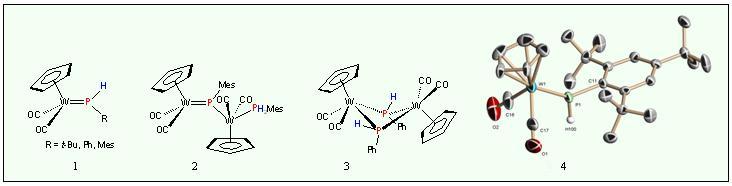
Despite
the notorious instability of most phosphenium complexes, a vast
chemistry has been established involving cycloaddition with organic
multiple bonded systems like isothiocyanates, dienes and diazoalkanes
followed in special cases by P-H-insertion to yield the coupling
products 5 – 8 .
- Organometallic Chemistry of Phosphorus Ylides (Publication Series 1 - 21)
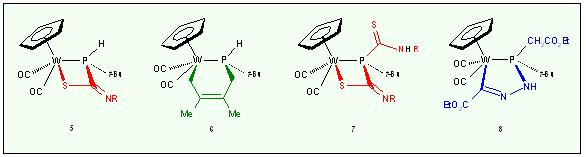
Reactivity of Metal Coordinated Carbonmonoxide towards Phosphorus ylides
This topic
investigates routes of C-C-coupling between CO coordinated to a metal
and strongly carbanionic phosphorus ylides. Starting with cationic
cyclopentadienyl carbonyl complexes opens up controlled interaction
involving nucleophilic attack at the CO carbon followed by
transylidation to yield metalloacyl-phhosphorusylides like 1. This
species develops ambidentate behavior towards carbon electrophiles
resulting in the formation of either the phosphonioacyl complex 2 or the phosphoniovinyl complex 3.
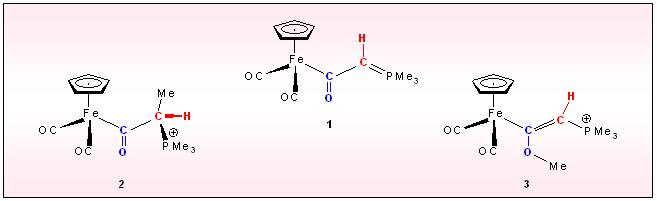
A special metall centered C-C coupling is initiated, when the silyl-methylene phosphorane iron speciec 4 is deprotonated. Consecutive C-C bond formation and transfer of the phophine to the metal generates the ketenyl complex 5. An analogous process is responsible for the formation of 6
starting from methyl carbonyl complexes of Mo, W and
trimethylmethylenephosphorane. The chelating ligand is composed of two
CO ligands the alkyl group and the ylidic carbon fragment.
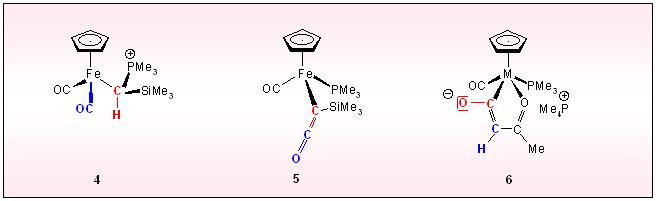
Carbonmonoxide cleaveage is achieved in the case of
tri(carbonyl)cyclopentadieny)manganese via the carbonmonoxide/ylide
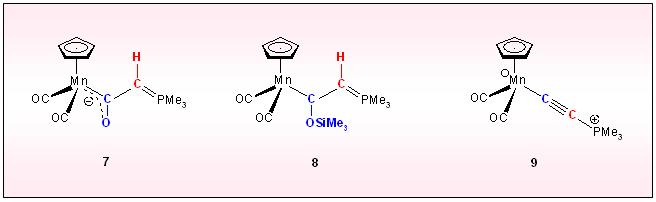
reaction to give the anionic acylylid complex 7, which on silylation converts to he siloxyvinyl complex 8. Clean silanol elimination on heating produces the phosphonioalkinyl complex 9.
Research Programmes
-
Sonderforschungsbereich 347: "Selektive Reaktionen Metall-aktivierter Moleküle" (1992 - 2002)
-
DFG-Schwerpunktsprogramm:
"Specific Phenomena in Silicon Chemistry: New Experimental and
Theoretical Approaches for the Controlled Formation and Better Understanding of Multidimensional Systems" (1996 - 2003)
- DFG-Schwerpunktsprogramm: "Phosphorchemie" (2000 - 2005)
Projects with Industry
Additional Activities
Basketball
- Organisation
Bundesliga-Basketball Würzburg. 2.Bundeslig Herren: 1989-1998. -
1.Bundesliga Herren: 1998-2005. - 2.Bundesliga Damen:1989-1996. -
1.Bundesliga Damen: 1996-1999.
- President 2.Bundesliga Herren: 1992-1998. - Vice-President 1.Bundesliga:1998-2004.
- During the Bundesliga Affiliation of the Würzburg Team the German National Players Dirk Nowitzki (Dallas Mavericks), Robert Garrett (Frankfurt Skyliners, Pompea Neapel, GHP Bamberg), Marvin Willoughby (Rheinergie Köln, Reggio Calabria, Pau-Orthez) and Desmond Green (Bayer Leverkusen, Alba Berlin) as well as the Nigerian National Player Olumide Oyedeji (Seattle Supersonics, Orlando Magics, Beijing Ducks) and the Croatian National Player Kresimir Loncar (Benetton Treviso, BC Kiev) have been scouted, educated and developed.
- In addition players of the Bundesliga-Teams were mainly responsible that the University of Würzburg won the German High School Championship in the years
- 1991, 1993, 1994, 1996, 1998 and 2001 for women and 1990, 1992, 1194, 1996, 1998 and 2000 for men.
- Participation in the League Cup TOP FOUR 1998 (Frankfurt) and 2004 (Frankfurt)
Honors: Carl-Diem-Medal for outstanding merits in sport
Music
- Leader of diverse bands: The Swinging Five, The Blizzards, The Nightbirds etc. (1962 -1975)